4 RNA-seq data analysis
4.1 What is differential gene expression?
Differential gene expression คือ การหาความแตกต่างของการแสดงออกของ gene ระหว่างกลุ่มตัวอย่างสองกลุ่มขึ้นไป เพื่อให้ได้ผลลัพธ์ว่ามี gene ตัวใดตัวหนึ่งแสดงออกมากหรือน้อยกว่าผิดปกติ เมื่อเทียบกับกลุ่มอื่นๆ โดยค่าที่นำมาใช้เปรียบเทียบนั้น จะได้มาจากขั้นตอน quantification
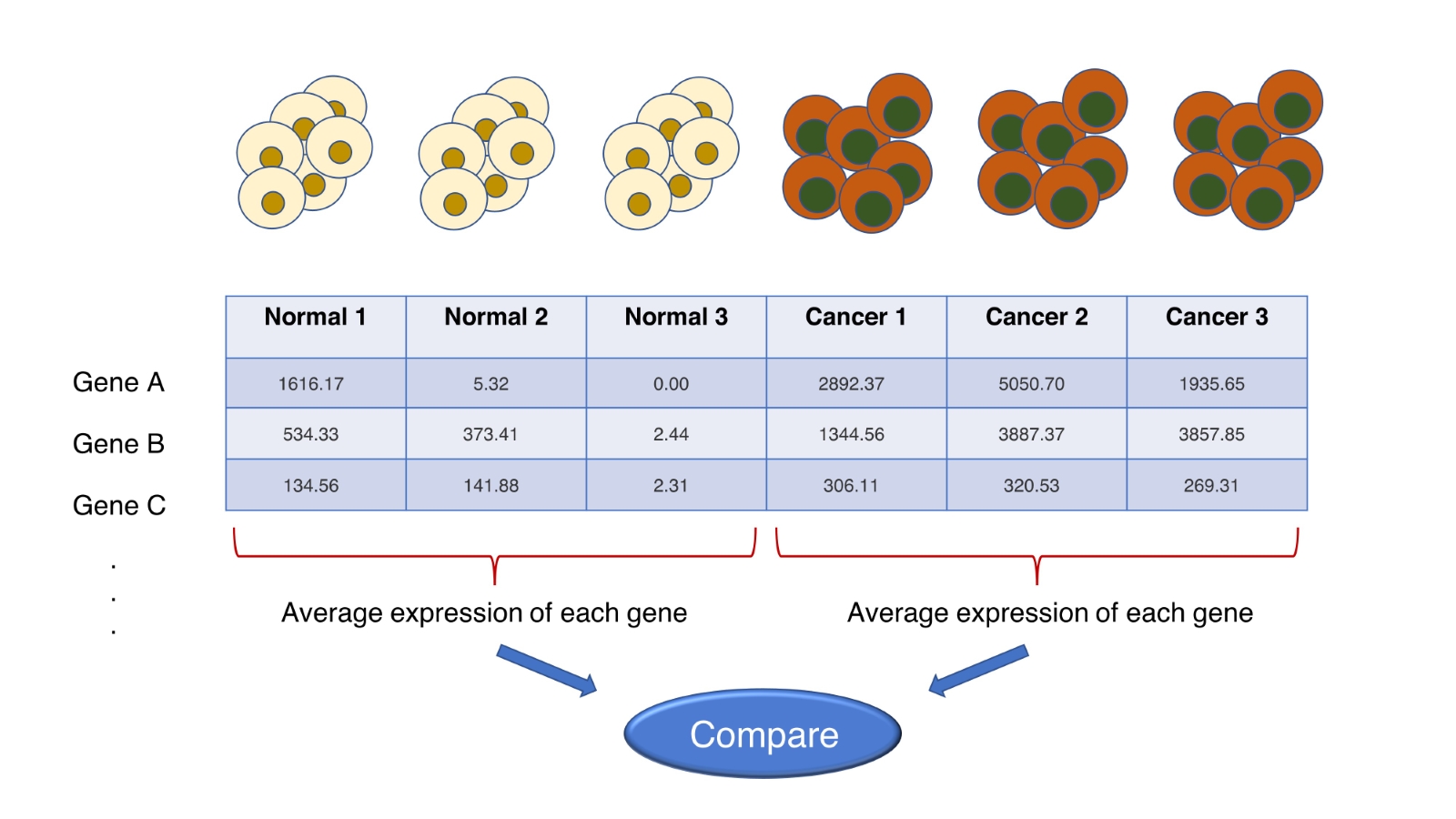
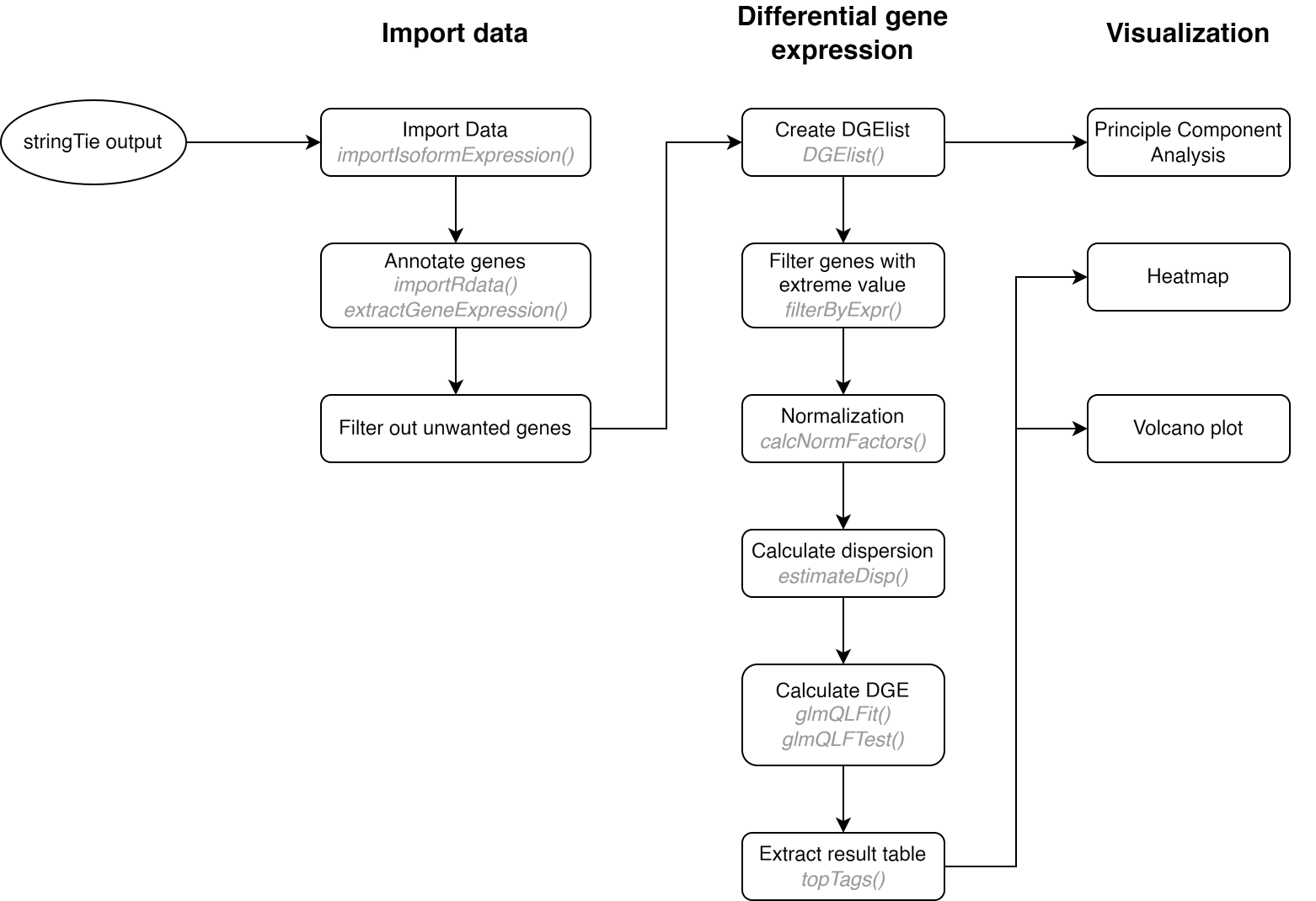
4.2 Differential gene expression workflow
หลังจากเราได้ข้อมูล Gene expression quantification จาก StringTie แล้ว เราจะนำข้อมูลมาผ่านกระบวนการต่างๆ เพื่อหาความแตกต่างของ gene แต่ละกลุ่ม
4.3 Import data
ในการที่จะนำไฟล์ RNA analysis เข้าสู่ R นั้น จำเป็นที่จะต้องเตรียมข้อมูลให้เหมาะกับ function ที่เราจะใช้ในการอ่านข้อมูล โดยในแต่ละ sample นั้น จะประกอบด้วยไฟล์ .gtf และ .ctab ที่ได้จากการวิเคราะห์ก่อนหน้านี้


4.3.1 Import quantification
โดยเราจะใช้ function importIsoformExpression ในการนำข้อมูลเข้าให้อยู่ในรูปของ dataframe
stringTie_quant <- importIsoformExpression(parentDir = "./Source/bladder", addIsofomIdAsColumn = FALSE,
readLength = 150)
head(stringTie_quant$counts, 10)จะเห็นว่าในไฟล์นั้นประกอบด้วย ส่วนแถว ซึ่งเป็นชื่อ isoform ของ RNA นั้นๆ และ ส่วนคอลัมน์ ซึ่งเป็นชื่อของ sample ที่เราศึกษา โดยข้อมูลแต่ละจุดคือ ค่าของ expression ที่ได้จากการวิเคราะห์
4.3.2 Make a design matrix
หลังจากนั้น เราต้องสร้าง condition matrix ซึ่งประกอบด้วย แต่ละ sample ที่ต้องการศึกษา และ condition ของตัวอย่างนั้น ซึ่งในที่นี้เราจะแบ่งเป็นสองกลุ่ม ก็คือ Normal และ Tumor
design <- data.frame(sampleID = colnames(stringTie_quant$abundance), condition = gsub(".{1}$",
"", colnames(stringTie_quant$abundance)) # Remove number
)
design4.3.3 Create a list of files
หลังจากนั้นเราจะต้องรวมไฟล์เข้ากันกับ annotation file ซึ่งจะทำการ annotate ชื่อ gene นั้น จาก Ensemble format เป็น gene id
switch_analyze_Rlist <- importRdata(isoformCountMatrix = stringTie_quant$counts,
isoformRepExpression = stringTie_quant$abundance, designMatrix = design, isoformExonAnnoation = "./Source/bladder/BCaMerge.gtf",
)##
|
| | 0%
|
|==================================================== | 50%
|
|=========================================================================================================| 100%
##
|
| | 0%
|
|=========================================================================================================| 100%
## comparison estimated_genes_with_dtu
## 1 N vs T 4239 - 7066names(switch_analyze_Rlist)## [1] "isoformFeatures" "exons" "conditions" "designMatrix"
## [5] "sourceId" "isoformCountMatrix" "isoformRepExpression" "runInfo"
## [9] "isoformRepIF"สังเกตว่าภายใน 1 list นั้นจะประกอบด้วยหลายหัวข้อ ซึ่งเราสามารถดึงออกมาใช้ได้ด้วย operator $
4.3.4 Extract gene count matrix
ต่อไปเราจะใช้แค่ gene count matrix จาก list ที่เราสร้างขึ้นมา
gene_count <- extractGeneExpression(switch_analyze_Rlist, extractCounts = TRUE # set to FALSE for abundances
)
head(gene_count, 10)จะเห็นว่าขณะนี้เรามีทั้ง gene_id และ gene_name แล้ว
4.3.5 Filter out lncRNA
ต่อไป เราจะนำรายชื่อของ RNA ที่เราไม่สนใจออกไป ซึ่งในที่นี้คือ long-noncoding RNA ซึ่งมักจะไม่ถูกเปลี่ยนไปเป็นโปรตีน แต่จะใช้สำหรับ function อื่นๆ ในร่างกาย
ก่อนอื่น เราต้อง import file ที่มีการ annotate ชนิดของ RNA เข้ามาใน R ก่อน โดยใช้ function rtracklayer::import()
V38.gtf <- rtracklayer::import("./Source/gencode.v38.annotation.gtf")
unique(V38.gtf$gene_type)## [1] "transcribed_unprocessed_pseudogene" "unprocessed_pseudogene" "miRNA"
## [4] "lncRNA" "protein_coding" "processed_pseudogene"
## [7] "snRNA" "transcribed_processed_pseudogene" "misc_RNA"
## [10] "TEC" "transcribed_unitary_pseudogene" "snoRNA"
## [13] "scaRNA" "rRNA_pseudogene" "unitary_pseudogene"
## [16] "polymorphic_pseudogene" "pseudogene" "rRNA"
## [19] "IG_V_pseudogene" "scRNA" "IG_V_gene"
## [22] "IG_C_gene" "IG_J_gene" "sRNA"
## [25] "ribozyme" "translated_processed_pseudogene" "vault_RNA"
## [28] "TR_C_gene" "TR_J_gene" "TR_V_gene"
## [31] "TR_V_pseudogene" "translated_unprocessed_pseudogene" "TR_D_gene"
## [34] "IG_C_pseudogene" "TR_J_pseudogene" "IG_J_pseudogene"
## [37] "IG_D_gene" "IG_pseudogene" "Mt_tRNA"
## [40] "Mt_rRNA"จะเห็นว่ามีชนิดของ RNA มากมายหลายชนิดในไฟล์นี้ เราจะทำการเลือกชื่อ RNA ที่เราไม่สนใจ ซึ่งก็คือ lncRNA มากรองข้อมูลในส่วนที่เราไม่ต้องการออกในไฟล์ต้นฉบับของเรา
หลังจากนั้นเราจะนำ column gene_id ออก และเปลี่ยน gene_name ให้เป็นชื่อแถว
Note: ในที่ข้อมูลนี้เราจะทำการตัด RNA ที่มีชื่อซ้ำออกไป เพื่อให้ง่ายแก่การสอน ซึ่งในการวิเคราะห์จริงอาจจะต้องใช้วิธีอื่นในการวิเคราะห์ชื่อ RNA ที่ซ้ำกันใน sample เดียวกัน
lncRNA_subset <- V38.gtf$gene_type == "lncRNA"
lncRNA <- V38.gtf[lncRNA_subset]$gene_name
gene_count_no_lncRNA <- gene_count %>% filter(!(gene_name %in% unique(lncRNA)))
head(gene_count_no_lncRNA, 10)## Get only count matrix
count_matrix <- gene_count_no_lncRNA %>%
distinct(gene_name, .keep_all = TRUE) %>% # Remove duplicate gene_name
column_to_rownames("gene_name") %>%
select(-gene_id) %>%
as.matrix
head(count_matrix, 10)## N1 N2 N3 T1 T2 T3
## TSPAN6 1616.16632 5.323296 0.000000 2892.36698 5050.6964 1935.65287
## DPM1 534.33260 373.409670 2.438447 1344.56193 3887.3732 3857.84694
## SCYL3 134.55807 141.880574 2.312303 306.11165 320.5272 269.31308
## C1orf112 68.44894 47.264863 17.260795 117.66245 207.5864 72.52807
## FGR 40.13006 3177.326075 62.927494 10.98451 192.8986 109.53769
## CFH 801.43355 8.169078 5.188639 1304.66135 13678.3377 2457.62369
## FUCA2 246.33240 104.995451 5.653375 1192.91176 3405.1256 1728.34830
## GCLC 374.82061 77.947080 6.817594 4565.35564 1549.1920 3010.92600
## NFYA 128.75396 215.173089 0.000000 526.52422 783.7322 664.80964
## STPG1 105.25642 27.359621 3.458716 253.40639 370.8472 179.81425เมื่อลองนำข้อมูลมาสร้าง boxplot คร่าวๆ จะพบว่ามีหลาย gene ที่มีความแตกต่างกัน ซึ่งต่อไปเราจะนำมาเข้าสู่กระบวนการหา differential gene expression เพื่อดูว่ามี gene ใดบ้างที่มีความแตกต่างกันระหว่างสองกลุ่มอย่างมีนัยสำคัญ
count_matrix %>%
edgeR::cpm(log = TRUE) %>%
head(20) %>%
t %>%
as.data.frame() %>%
rownames_to_column("type") %>%
tidyr::pivot_longer(-type) %>%
mutate(type = gsub("\\d", "", type)) %>%
ggplot(aes(x = name, y = value, fill = type)) + geom_boxplot() + theme_bw() +
theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust = 1))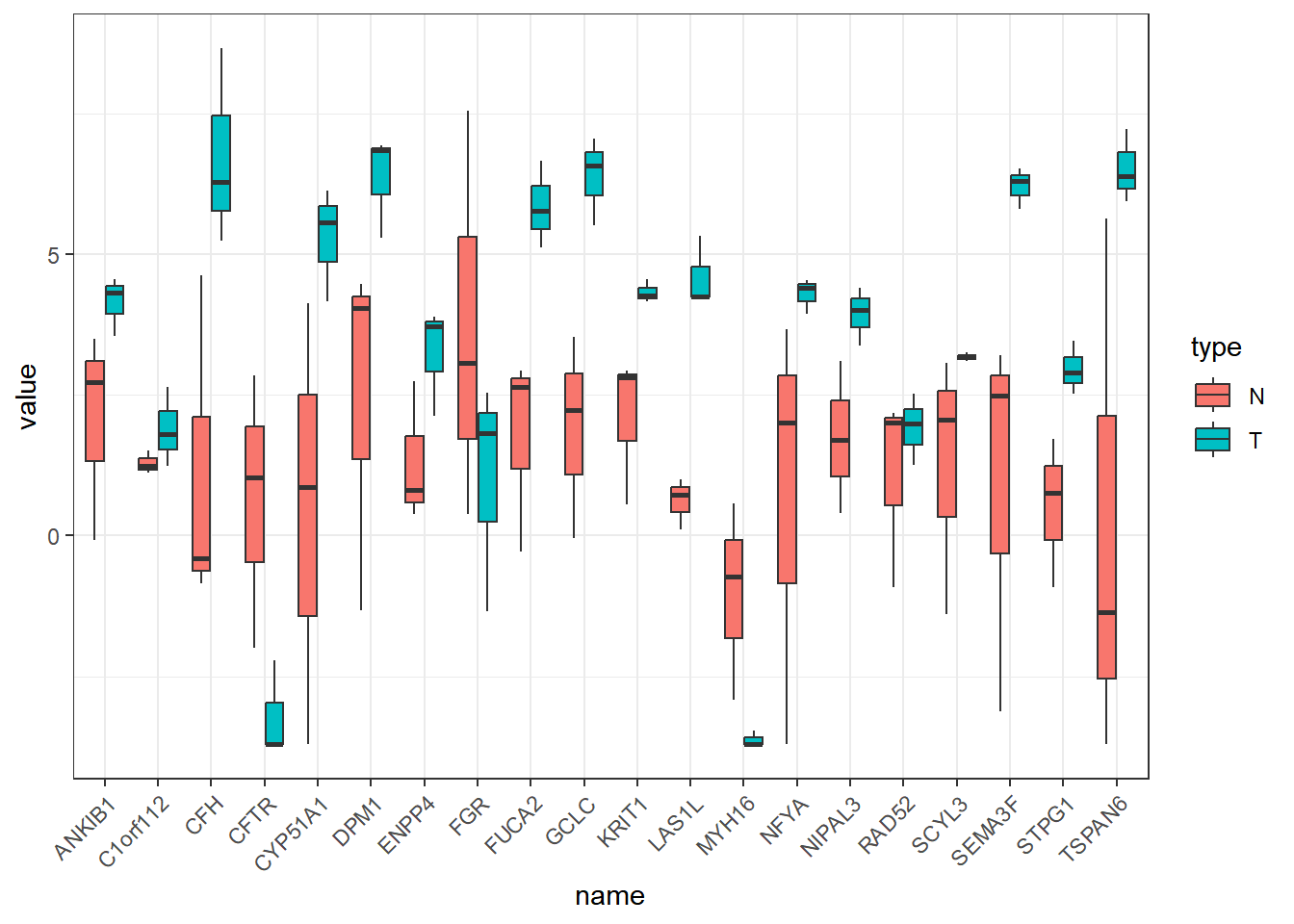
4.4 Differential gene expression analysis
ก่อนที่เราจะทำการ visualize ข้อมูลนั้น เราจะต้องทำการวิเคราะห์ก่อนว่า RNA ไหนที่มีการแสดงออกระหว่างสองกลุ่มที่แตกต่างอย่างมีนัยสำคัญ
โดยเราจะเริ่มจากการสร้าง design matrix ซึ่งบ่งบอกว่าใครอยู่กลุ่มไหน
library(edgeR)
colnames(count_matrix)## [1] "N1" "N2" "N3" "T1" "T2" "T3"group <- design$condition
diff_design <- model.matrix(~0 + group)
diff_design## groupN groupT
## 1 1 0
## 2 1 0
## 3 1 0
## 4 0 1
## 5 0 1
## 6 0 1
## attr(,"assign")
## [1] 1 1
## attr(,"contrasts")
## attr(,"contrasts")$group
## [1] "contr.treatment"สิ่งที่เราเห็นคือ design matrix ของกลุ่มที่เราต้องการ โดยหมายเลข 1 คือตัวบ่งบอกว่า sample เราอยู่ในกลุ่มนั้นๆ โดยในที่นี่ sample 1-3 จะอยู่ในกลุ่ม Normal ส่วน sample 4-6 จะอยู่ในกลุ่ม Tumor
4.4.1 Normalization
หลังจากนั้น เราจะต้องทำการ normalize ค่าการแสดงออกของ RNA เนื่องจากการ run RNA seq ในแต่ละ sample นั้น สภาวะของเครื่องอาจจะมีความแตกต่างกันบ้างเล็กน้อย ส่งผลให้ค่า signal intensity พื้นหลังนั้นมีไม่เท่ากัน
dge <- DGEList(counts = count_matrix, group = group)
keep <- filterByExpr(dge, group = group, min.count = 2, min.prob = 0.5)
dge <- dge[keep, ]
# Calculate normalization factor
genexp <- calcNormFactors(dge)
# GLM Common dispersion
genexp <- estimateGLMCommonDisp(dge, diff_design)
# Estimate GLM trended dispersions
genexp <- estimateGLMTrendedDisp(genexp, diff_design)
# Tagwise dispersion of each gene
genexp <- estimateGLMTagwiseDisp(genexp, diff_design)
plotBCV(genexp)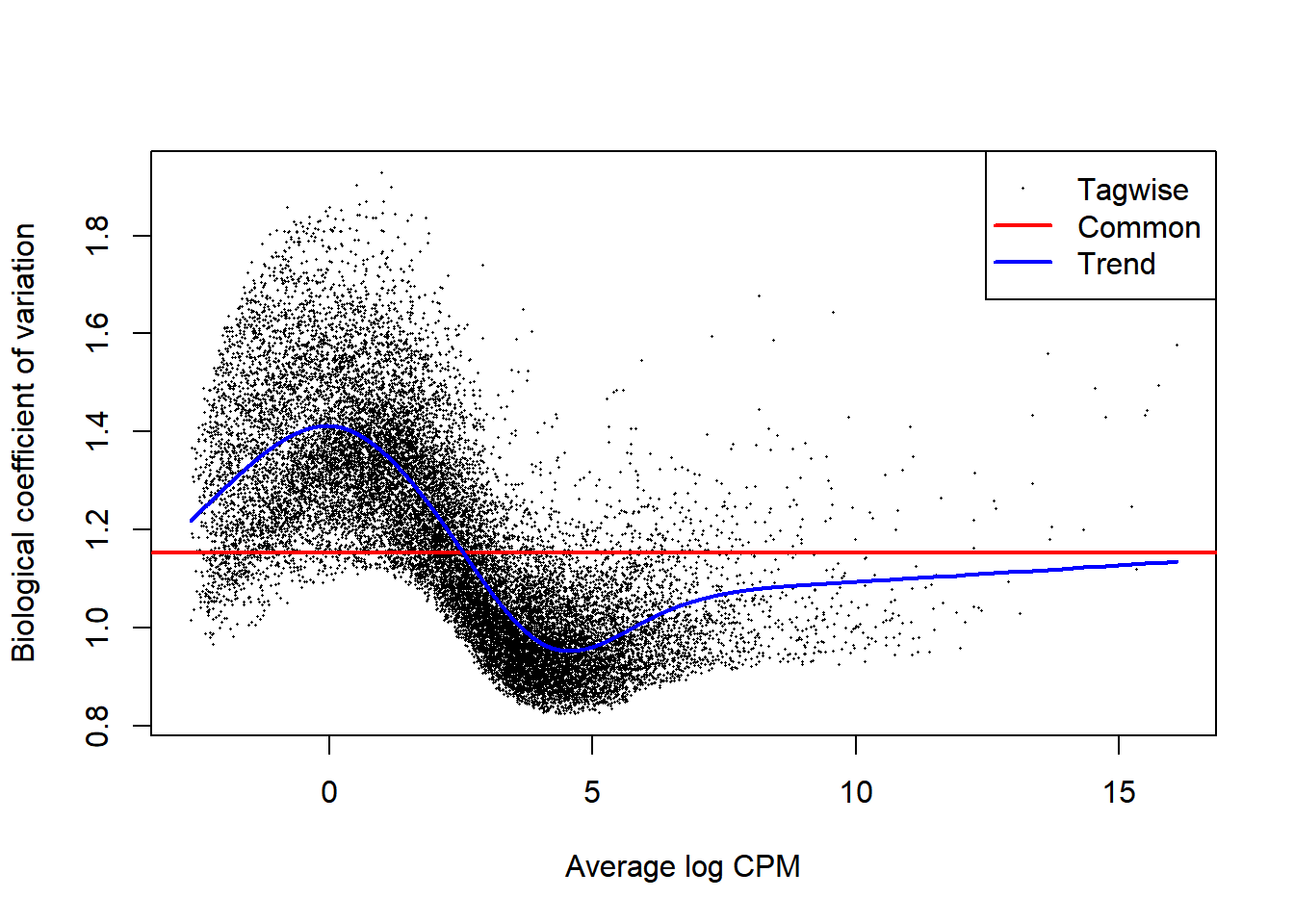
head(genexp$counts)## N1 N2 N3 T1 T2 T3
## TSPAN6 1616.16632 5.323296 0.000000 2892.36698 5050.6964 1935.65287
## DPM1 534.33260 373.409670 2.438447 1344.56193 3887.3732 3857.84694
## SCYL3 134.55807 141.880574 2.312303 306.11165 320.5272 269.31308
## C1orf112 68.44894 47.264863 17.260795 117.66245 207.5864 72.52807
## FGR 40.13006 3177.326075 62.927494 10.98451 192.8986 109.53769
## CFH 801.43355 8.169078 5.188639 1304.66135 13678.3377 2457.62369หลังจาก normalize แล้ว เราจะทำการวิเคราะห์ differential gene expression โดยการใช้การวิเคราะห์ทางสถิติที่เรียกว่า negative binomial generalized log-linear model ซึ่งโดยสรุปคร่าวๆ คือการเปรียบเทียบ average log RNA expression ระหว่างสองกลุ่ม แต่ซับซ้อนกว่าเพื่อลด ผลบวกลวง
Note: package ที่นิยมใช้ในปัจจุบัน ได้แก่ limma, edgeR, และ DEseq โดยจะมีความแตกต่างกันเล็กน้อยในส่วนของการวิเคราะห์ทางสถิติ สำหรับผู้ที่สนใจสามารถศึกษาเพิ่มเติมได้ที่ https://www.biostars.org/p/284775/
หลังจากนั้นเราจะใช้ funtion topTags() เพื่อทำการดึงตารางผลของ differential RNA expression ออกมา
fit <- glmQLFit(genexp, diff_design)
genediff <- glmQLFTest(fit, contrast = c(-1, 1))
# All genes
all_gene <- topTags(genediff, n = Inf, p.value = 1, adjust.method = "fdr")
# Only significant value
sig_gene <- topTags(genediff, n = Inf, p.value = 0.05, adjust.method = "fdr", sort.by = "logFC")
# Total differentiated gene
summary(decideTests(genediff))## -1*groupN 1*groupT
## Down 1792
## NotSig 17504
## Up 939# Summary table
sig_gene$tableโดยจากตาราง จะพบว่ามีการแสดงค่าต่างๆ โดยที่เราสนใจมักจะเป็น
- logFC ซึ่งก็คือ fold change ของ RNA expression ระหว่างกลุ่ม Normal vs Tumor
- pvalue โดยเรามักจะต้องปรับผลเพื่อลดภาวะผลบวกลวงออกไปด้วย เราจึงใช้ column FDR ไม่ใช่ PValue
4.5 Visualization
4.5.1 Principal Component Analysis (PCA)
PCA คือการลดมิติของปริมาณข้อมูลลงเพือทำให้เกิดความง่ายขึ้นในการวิเคราะห์ โดยใช้หลักการรวมข้อมูลแบบ linear combination ที่มีความแปรปรวนใกล้เคียง ซึ่งโดยทั่วไปแล้ว จะนำมาใช้ในการดูความแตกต่างกันของลักษณะข้อมูลในแต่ละกลุ่มแบบคร่าวๆ หรือใช้ในการค้นหาความผิดปกติของข้อมูลที่เกินจากสภาวะที่ต่างกัน (batch effect) โดยที่ข้อมูลที่มีลักษณะใกล้เคียงกันจะอยู่ในตำแหน่งที่ใกล้เคียงกัน
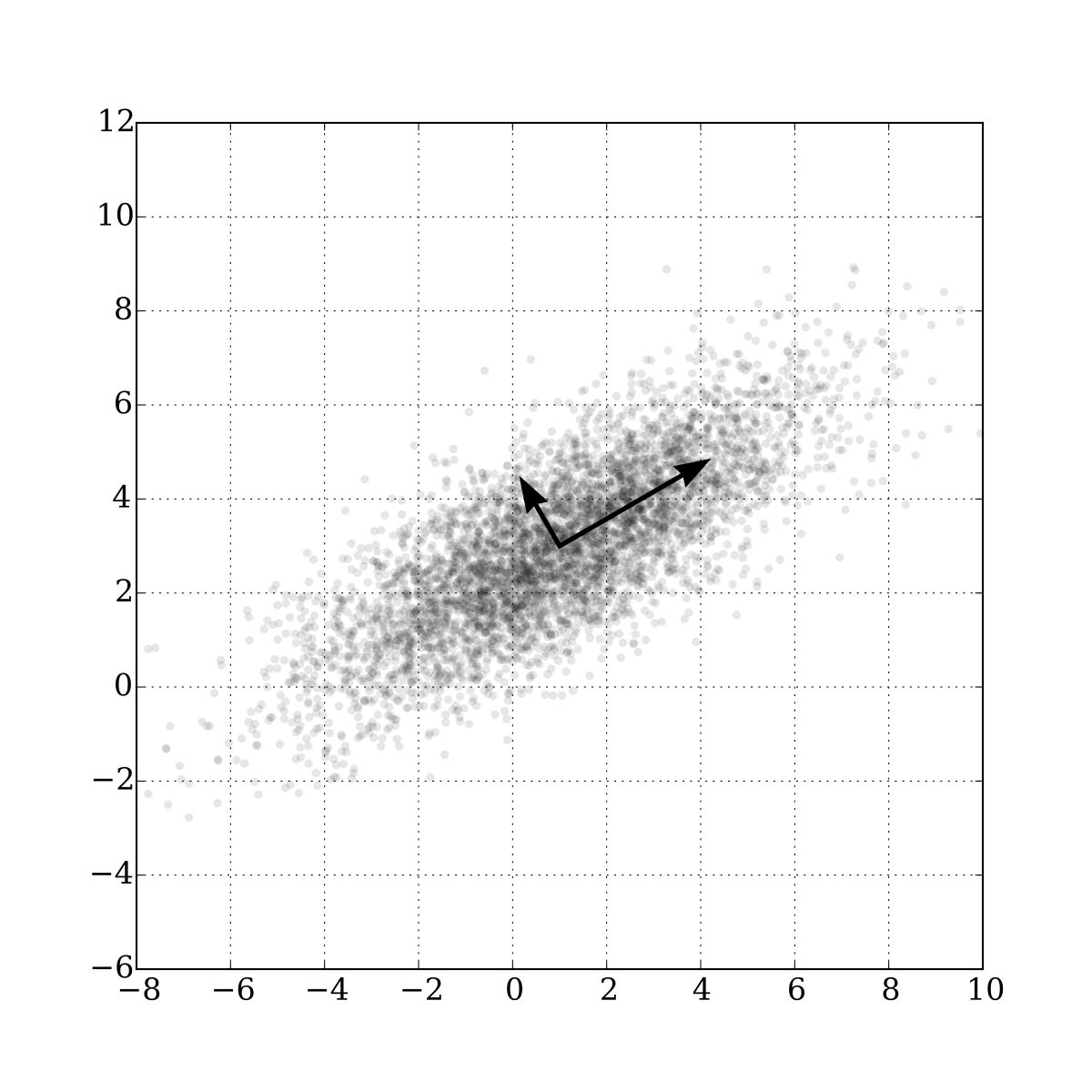
รูปจาก: https://en.wikipedia.org/wiki/Principal_component_analysis
4.5.1.1 Requirement
- ข้อมูลควรมีการถูก normalized โดยอาจจะ centered (ทำให้ scale เริ่มต้นที่ 0) หรือไม่ก็ได้
- ต้องไม่มี missing value
library(PCAtools)
# Calculate log-counts-per-million
logcpm <- cpm(dge, prior.count = 2, log = TRUE)
# Create a metadata table
metadata <- data.frame(row.names = colnames(logcpm), group = c(rep(1, 3), rep(2,
3)))
metadataโดยการแปลผล PCA นั้น ควรดูไล่ไปทีละแกน (มิติ 1 -> มิติ 2 ไม่ใช่ดู 2 มิติพร้อมกัน)
# Perform PCA analysis
pc <- pca(logcpm, metadata = metadata, removeVar = 0.1)
# Create PCA plot
biplot(pc, colby = "group")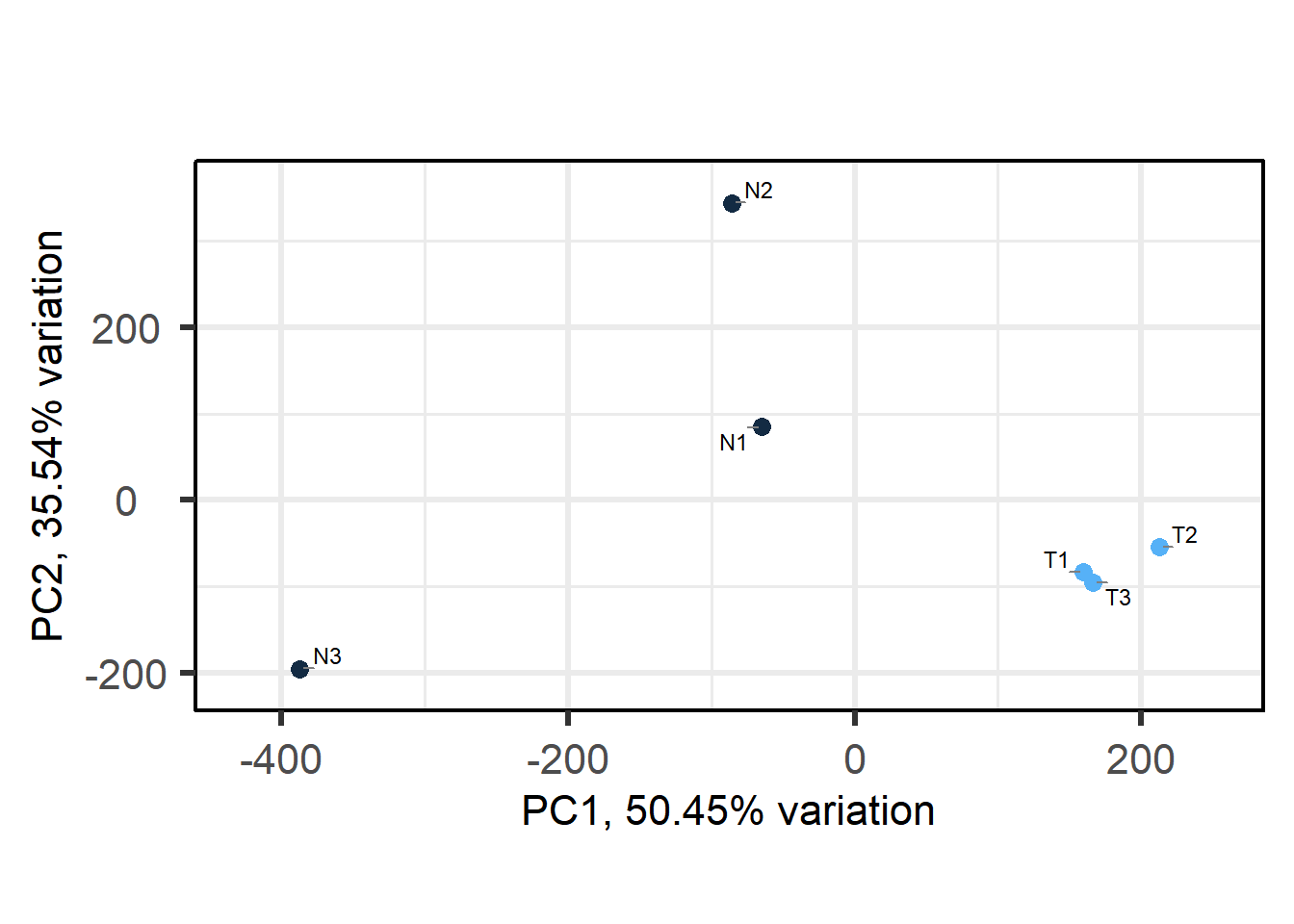
จะเห็นได้ว่า ในส่วนของ T1, T2 และ T3 นั้นค่อนข้างเกาะกลุ่มกัน แต่ N นั้น มีความแตกต่างกันพอสมควรในทั้งสองมิติ
แม้ว่าในกราฟจะมีแค่ 2 มิติ แต่โดยที่จริงแล้วมิตินั้นจะโดนลดลงเหลือ n มิติ
pairsplot(pc)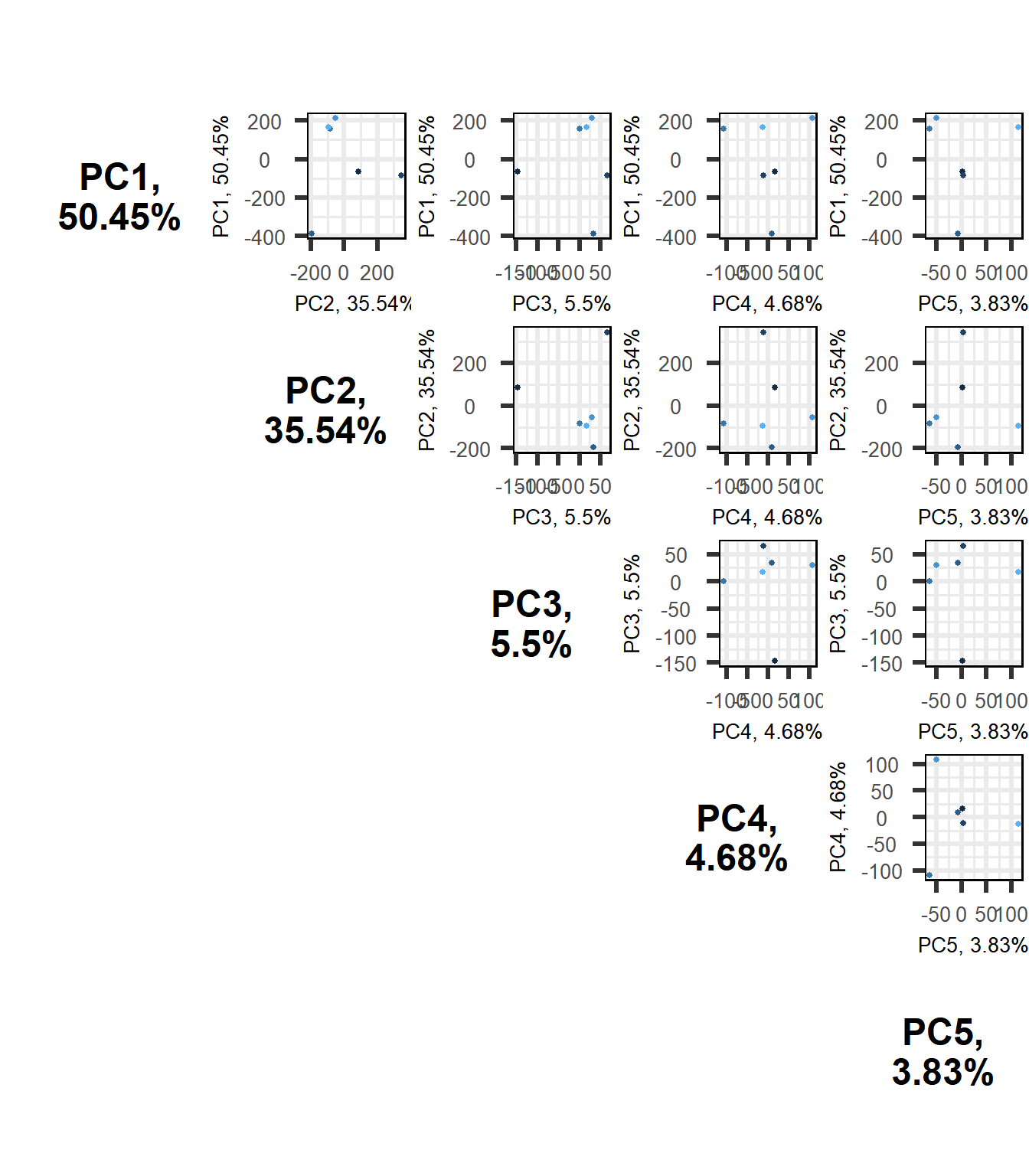
ซึ่งเราสามารถดูความมากน้อยของผลกระทบของในแต่ละมิติได้โดยใช้ Scree plot โดยมิติแรกจะมีผลมากกว่ามิติหลังเสมอ
screeplot(pc)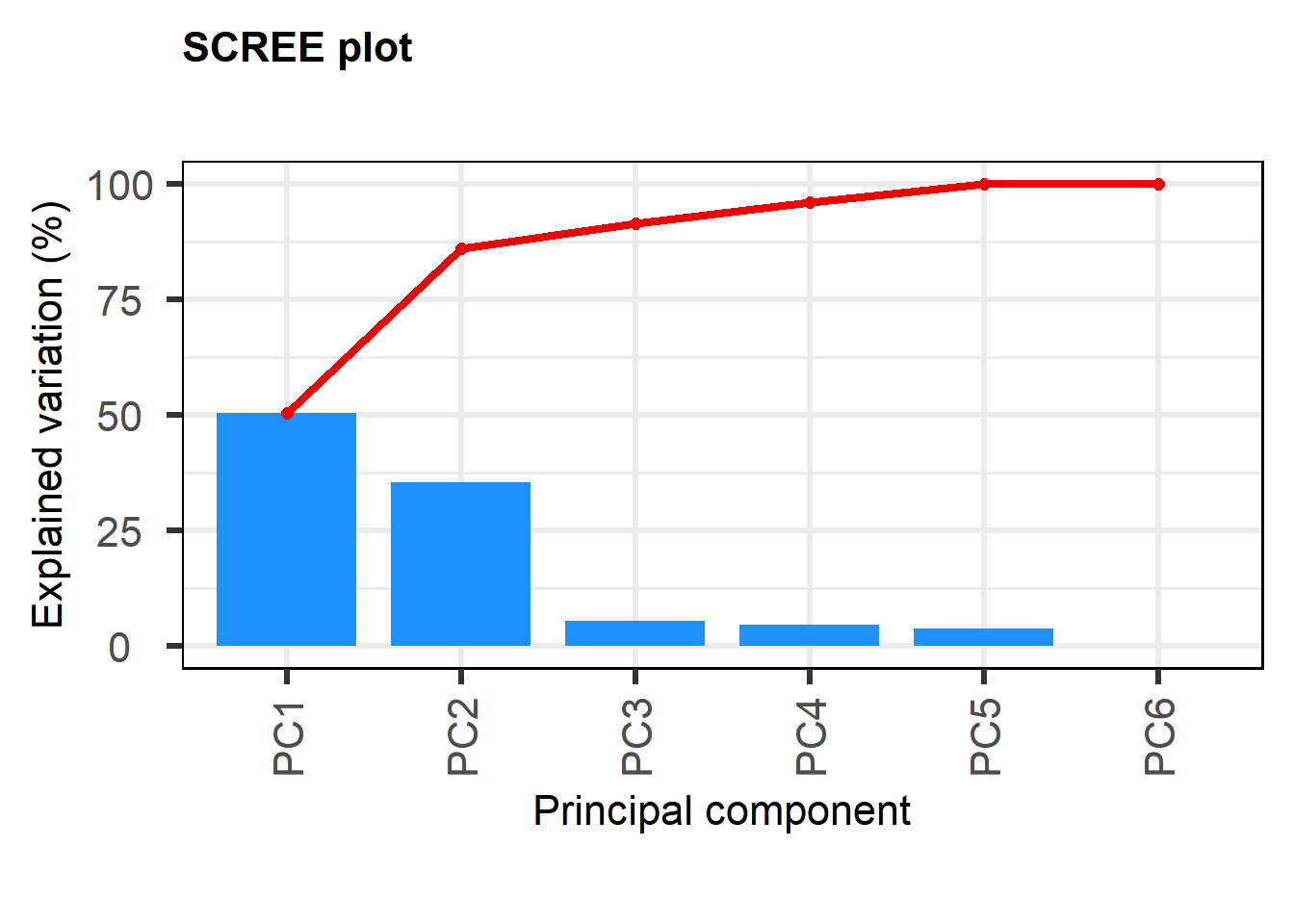
ในส่วนของข้อมูลเชิงลึกของ PCA สามารถศึกษาเพิ่มเติมได้ในเอกสารแนบ: http://www.cs.otago.ac.nz/cosc453/student_tutorials/principal_components.pdf
ตัวอย่างการใช้งานเพิ่มเติม: https://www.bioconductor.org/packages/release/bioc/vignettes/PCAtools/inst/doc/PCAtools.html
4.5.2 Heatmap
Heatmap คือการเปลี่ยนข้อมูลที่มีให้อยู่ในรูปของสี ซึ่งจะแสดงความแตกต่างตามค่าที่มากหรือน้อย โดยการสร้าง heatmap นั้นจะใช้ข้อมูลดิบ (ก่อนทำ differential expression) ซึ่งจะทำให้เห็นภาพรวมของข้อมูลแต่จะไม่ให้ข้อมูลความแตกต่างทางด้านสถิติมากนัก
ซึ่งโดยปกติถ้านำข้อมูลทั้งหมดมาสร้าง heatmap จะทำให้รูปมีขนาดใหญ่เกินไป ดังนั้น เรามักจะกรองข้อมูลที่เราต้องการจะนำเสนอก่อนที่จะนำมาสร้าง
library(ComplexHeatmap)
# Filter only significant DEGs gene (from EdgeR)
DEGGene <- logcpm[rownames(sig_gene$table), ]
Heatmap(DEGGene, row_km = 6, name = "RNAseq\nWorkshop", row_names_gp = gpar(fontsize = 3))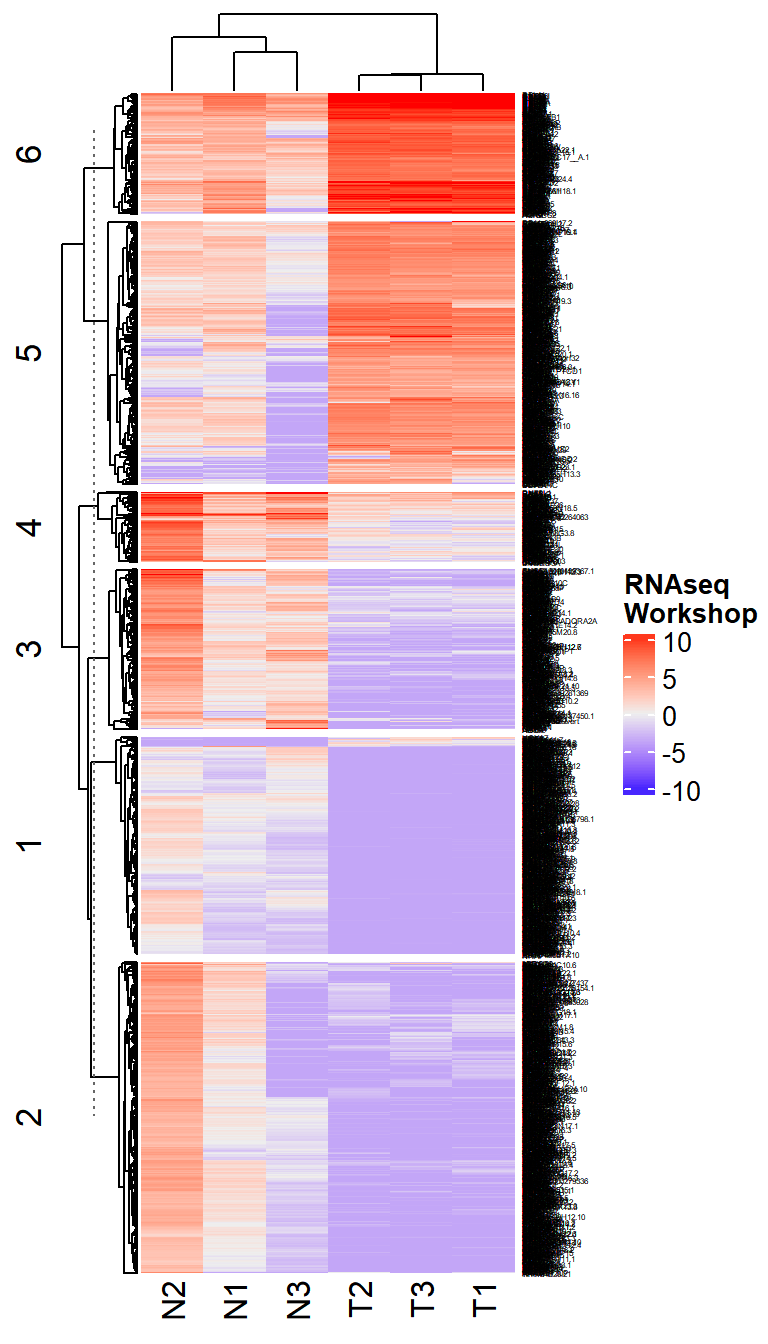
ตัวอย่างการใช้งานเพิ่มเติม: https://jokergoo.github.io/ComplexHeatmap-reference/book/
4.5.3 Volcano plot
Volcano plot คือกราฟที่แสดงความแตกต่างของการแสดงออกของ RNA ระหว่างสองกลุ่ม โดยมีแกน x คือ log fold change และ y คือ -log10(p-value) เหตุผลที่แกน y ต้องเป็น -log10(p-value) เพื่อที่จะปรับค่า p-value ที่เป็นทศนิยมนั้นให้อยู่ในหลักจำนวนเต็ม ซึ่งจะทำให้ได้กราฟที่มีรูปร่างคล้ายภูเขาไฟหัวกลับ
library(EnhancedVolcano)
# Create Volcano plot
EnhancedVolcano(all_gene$table, lab = rownames(all_gene$table), x = "logFC", y = "PValue",
xlim = c(-12, 12), labSize = 2, pCutoff = 0.05, title = "RNAseq workshop", max.overlaps = 50)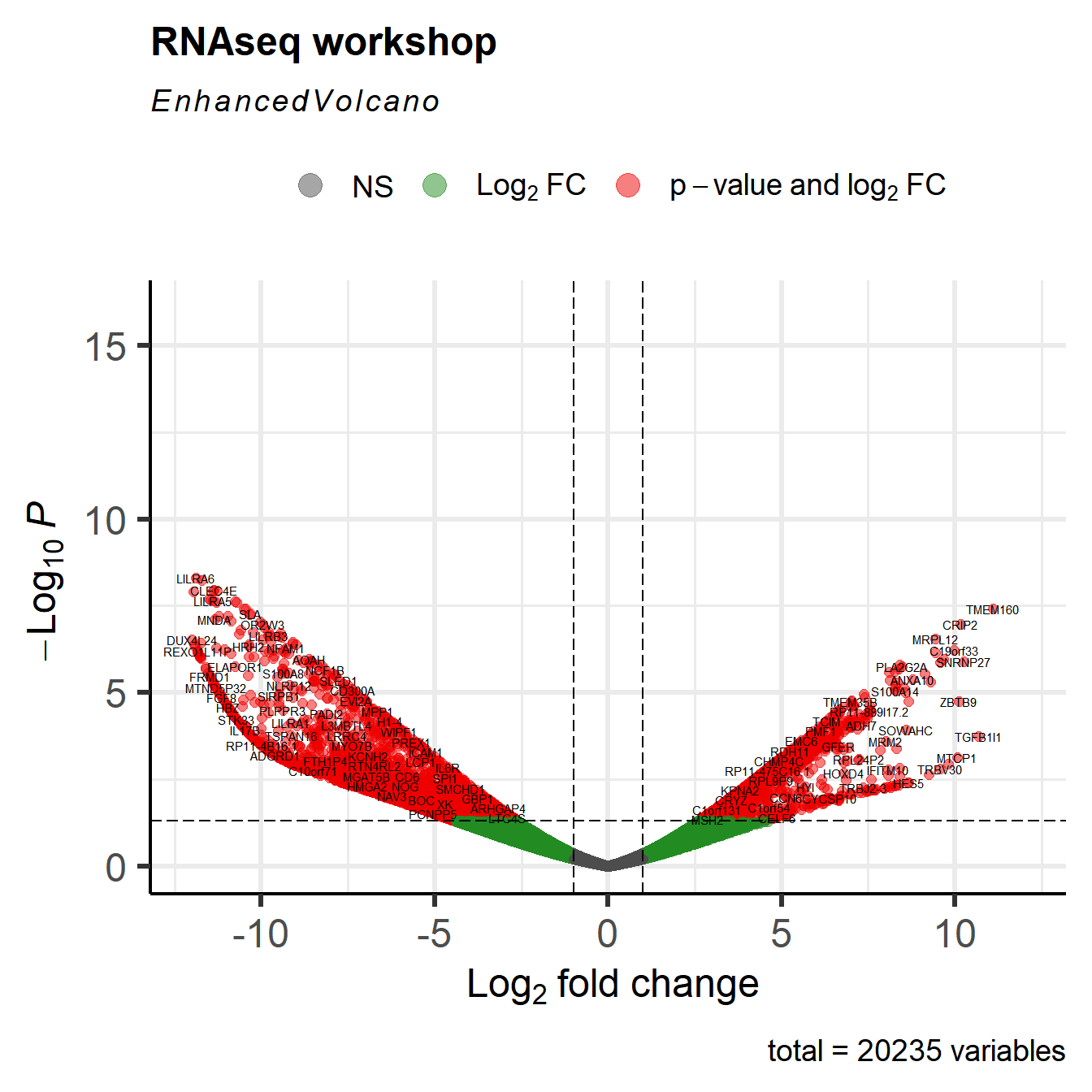
ค่าที่ cut-off ที่เราสนใจนั้นมักจะเป็นที่ logFC > 1-2, และ -log10(p-value) > 1.3-2 (p-value < 0.01-0.05)
ตัวอย่างการใช้งานเพิ่มเติม: https://bioconductor.org/packages/release/bioc/vignettes/EnhancedVolcano/inst/doc/EnhancedVolcano.html